Insights
Simple scalable graph neural networks
By
and
Monday, 19 April 2021
One of the challenges that has prevented the wide adoption of graph neural networks in industrial applications is the difficulty to scale them to large graphs, such as Twitter’s social network. The interdependence between nodes makes the decomposition of the loss function into individual node contributions challenging. In this post, we describe a graph neural network architecture (SIGN) that is of simple implementation and that works on very large graphs, thus being particularly suited to large-scale industrial applications, such as Twitter.
Graph Neural Networks (GNNs) are a class of machine learning models that have emerged in recent years for learning on graph-structured data. GNNs have been successfully applied to model systems of relation and interactions in a variety of domains, such as social science, chemistry, and medicine. Until recently, most of the research in the field has focused on developing new GNN models and testing them on small graphs¹ and relatively little effort has been invested in dealing with large-scale applications. However, industry problems often deal with giant graphs. Twitter’s social network contains hundreds of millions of nodes and billions of edges and most methods described in the literature are unsuitable for these settings.
GNNs can be designed to make predictions at the level of nodes, edges, or entire graphs. For example, a prediction at a node level could be for applications such as detecting malicious actors online. An edge-wise prediction task could be link prediction, which is a typical scenario in recommender systems. A graph-wise prediction task could be predicting chemical properties of molecular graphs.
In a nutshell, GNNs work by aggregating the features from local neighbor nodes. You can arrange node features into a nxd matrix, X (here n denotes the number of nodes and d denotes the number of input features)² and combine node-wise transformations with feature diffusion across adjacent nodes as:
Y = ReLU(AXW)
In the above definition, W is a learnable matrix shared across all nodes, A is a linear diffusion operator amounting to a weighted average of features in a neighborhood³, and ReLU (Rectified Linear Unit) applies the nonlinear activation ReLU(x)= max(0, x).
This formulation is the simplest convolution-like operation on graphs, implemented in the popular graph convolution network (GCN) model. Multiple layers of this form can be applied in sequence like in traditional convolutional neural networks (CNNs). For instance, the node-wise classification task, the one that we focus on in this post, can be carried out by a two-layer GCN model of the form:
Y = softmax(A ReLU(AXW) W’)
Why is scaling GNNs challenging? In traditional machine learning settings, it is typically assumed that the samples are drawn from some distribution in a statistically independent manner. This assumption allows you to decompose the loss function into the individual sample contributions. It also allows you to use stochastic optimization techniques which work with small subsets (mini-batches) of the training data at a time. Nowadays, virtually every deep neural network architecture is trained using mini-batches.
However, in the above node classification task, samples are the nodes that the GNN is trained on, and the following observations need to be considered:
In many early works on graph neural networks, the above problems were ignored. Architectures such as GCN and ChebNet², MoNet⁴ and GAT⁵ were trained using full-batch gradient descent, which holds the entire graph adjacency matrix and node features in memory. As a result, for example, if E represents the cardinality of the graph edge set, a L-layer GCN model has time complexity 𝒪(LEd + Lnd²) and memory complexity 𝒪(Lnd +Ld²), which makes the use of these architectures prohibitive even for modestly-sized graphs⁷.
The first work to tackle the problem of scalability was GraphSAGE⁸, a seminal paper of Will Hamilton and co-authors. GraphSAGE used neighborhood sampling combined with mini-batch training to train GNNs on large graphs. The main observation is that, to compute the training loss on a single node with an L-layer GCN, only the L-hop neighbors of that node are necessary, as nodes further away in the graph are not involved in the computation.
The problem is that, for graphs of the “small-world” type, such as social networks, the 2-hop neighborhood of some node might already contain millions of nodes, making it too big to be stored in memory⁹. GraphSAGE tackles this problem by sampling the neighbors up to the L-th hop. Starting from the training node, it samples uniformly with replacement¹⁰ a fixed number k of 1-hop neighbors, then for each of these neighbors it again samples k neighbors, and so on for L times. In this way, for every node we are guaranteed to have a bounded L-hop sampled neighborhood of 𝒪(kL) nodes. If we then construct a batch with b training nodes, each with its own independent L-hop neighborhood, we get to a memory complexity that is independent of the graph size n: 𝒪(bkL). The computational complexity of one batch of GraphSAGE is 𝒪(bLd²kL).
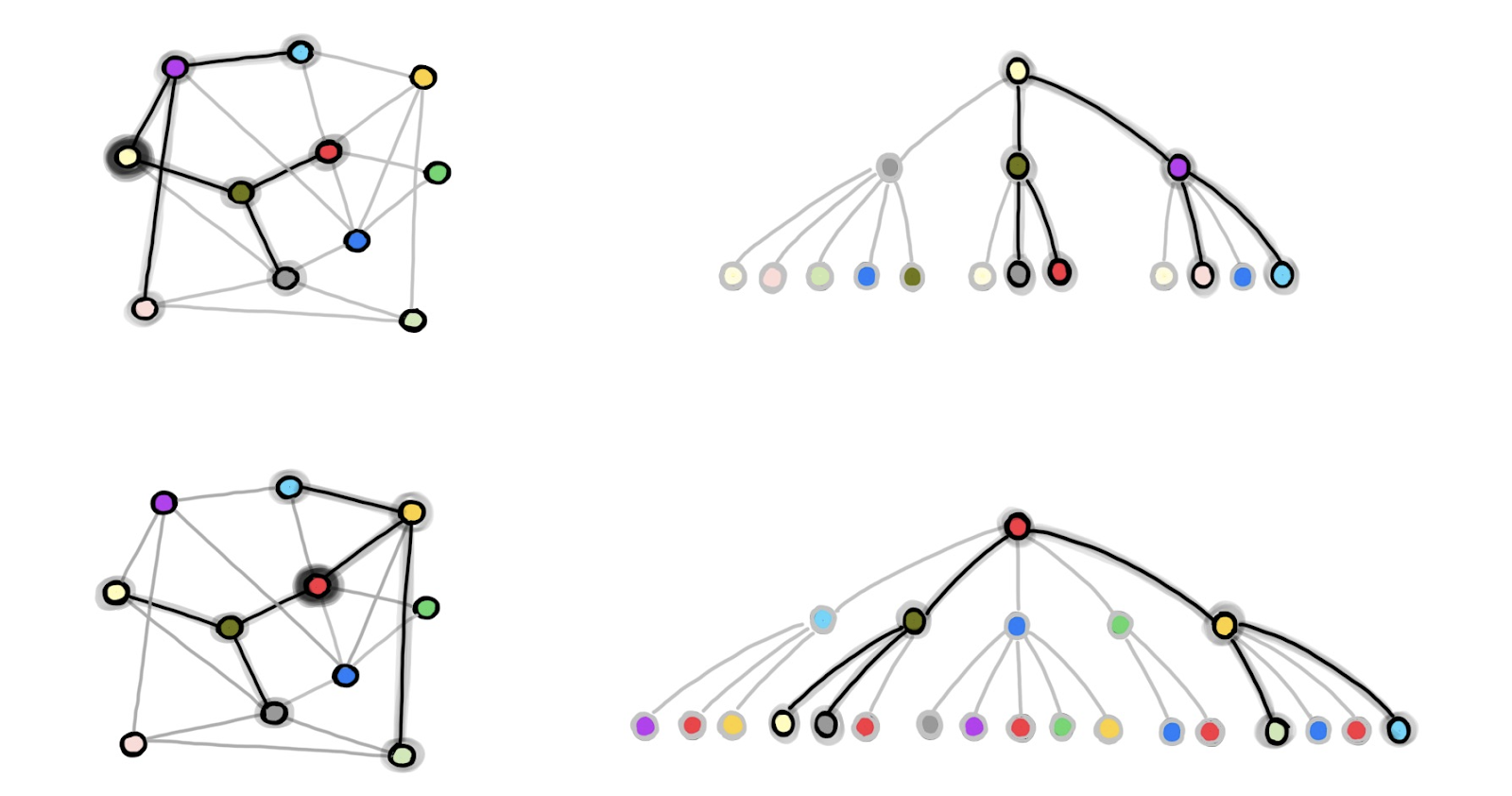
Figure 1 shows an example of neighborhood sampling procedure of GraphSAGE. On the left, a batch of b nodes is subsampled from the full graph (in this example, b=2 and the red and light yellow nodes are used for training). On the right, the 2-hop neighborhood graphs sampled with k=2, which are independently used to compute the embedding and therefore the loss for the red and light yellow nodes.
A notable drawback of GraphSAGE is that sampled nodes might appear multiple times, thus potentially introducing a lot of redundant computation. For instance, in the figure above the olive green node appears in both the 1-hop neighborhoods of the two (light yellow and red) training nodes. Therefore, its embedding is computed twice in the batch. With the increase of the batch size b and the number of samples k, the amount of redundant computation increases as well. Moreover, despite having 𝒪(bkL)nodes in memory for each batch, the loss is computed on only b of them, and therefore, the computation for the other nodes is also somewhat wasted.
Multiple follow-up works focused on improving the sampling of mini-batches to remove redundant computation of GraphSAGE and make each batch more efficient. The most recent works in this direction are ClusterGCN¹¹ and GraphSAINT¹², which take the approach of graph-sampling (as opposed to neighborhood-sampling of GraphSAGE). In graph-sampling approaches, for each batch, a subgraph of the original graph is sampled, and a full GCN-like model is run on the entire subgraph. The challenge is to make sure that these subgraphs preserve most of the original edges and still present a meaningful topological structure.
ClusterGCN achieves this by first clustering the graph. Then, at each batch, the model is trained on one cluster. This allows the nodes in each batch to be as tightly connected as possible.
GraphSAINT proposes instead a general probabilistic graph sampler that constructs training batches by sampling subgraphs of the original graph. The graph sampler can be designed according to different schemes. For example, it can perform uniform node sampling, uniform edge sampling, or “importance sampling” by using random walks to compute the importance of nodes and use it as the probability distribution for sampling.
An advantage of sampling is that, by randomly dropping a fraction of edges during training, it acts as a sort of edge-wise dropout, which regularizes the model and possibly improves performance¹³. However, edge-wise dropout still requires you to see all the edges at inference time, which is not feasible when there are too many. Graph sampling might also reduce the bottleneck¹⁴ and the resulting “over-squashing” phenomenon that stems from the exponential expansion of the neighborhood.
It is our belief, however, that graph-sampling is not the ultimate solution to build scalable GNNs. The above positive and negative effects of graph-sampling have not been investigated properly. The positive effects, other than alleviating computational complexity, are not clear. Additionally, the extent of the negative effects, including implementation complexities and bias, have not been comprehensively accounted for or measured. It is for this reason that we believe creating a simple, strong, sampling-free, and scalable baseline architecture remains a meaningful and appealing pursuit.
In our recent paper with Ben Chamberlain, Davide Eynard, Emanuele Rossi, and Federico Monti¹⁵, we investigate the extent to which it is possible to design simple, sampling-free architectures for node-wise classification problems.
Our approach is motivated by several recent empirical findings. First, Shchur and coauthors have shown that in node-wise classification tasks, simple fixed aggregators (such as GCN) often outperform more complex ones¹⁶ (such as GAT or MPNN⁶). Second, while the success of deep learning was built on models with a lot of layers, in graph deep learning it is still an open question whether depth is needed. In particular, Wu and coauthors¹⁷ argue that a GCN model with a single multi-hop diffusion layer can perform on par with models with multiple layers.
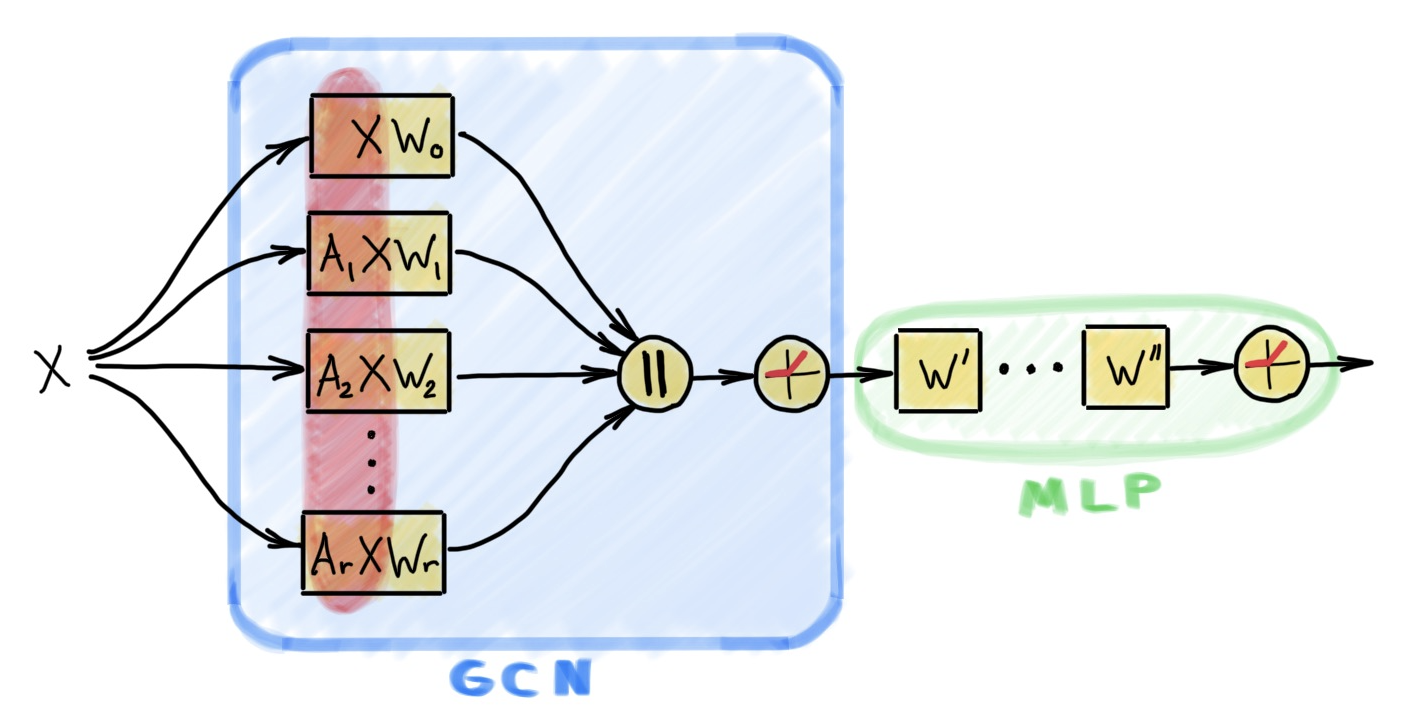
The important observation here is that a model with only one single convolutional layer has the remarkable advantage that all the graph-related (fixed) operations are in the first layer of the architecture and can therefore be precomputed. The pre-aggregated information can then be fed as inputs to the rest of the model. Due to the lack of neighborhood aggregation in the model, this model boils down to a multi-layer perceptron (MLP) that can be trained with standard mini-batch gradient descent. We increase the expressivity of such a model by combining several, possibly specialized and more complex, diffusion operators within a single convolutional layer. In this way, it is possible not only to obtain an extremely scalable, samping-free model, but to also retain enough model capacity to compete for state-of-the-art results against deeper models¹⁸. Examples of specialized operators include those accounting for local substructure counting¹⁹ or graph motifs²⁰.
The proposed scalable architecture, which we call Scalable Inception-like Graph Network (SIGN) has the following form for node-wise classification tasks:
Y = softmax(ReLU(XW₀ | A₁XW₁ | A₂XW₂ | ... | AᵣXWᵣ) W’)
In the above definition, Aᵣ are linear diffusion matrices (such as a normalized adjacency matrix, its powers, or a motif matrix), Wᵣ and W’ are learnable parameters, and | refers to concatenation.
As depicted in Figure 2, the network can be made deeper with additional node-wise layers,
Y = softmax(ReLU(...ReLU(XW₀ | A₁XW₁ | A₂XW₂ | ... | AᵣXWᵣ) W’)... W’’)
Finally, when employing different powers for the same diffusion operator (for example, A₁=B, A₂=B², A₃=B³), the graph operations effectively aggregate from neighbors in further and further hops towards multiscale neighborhood representations. This is similar to having convolutional filters of different receptive fields within the same network layer. This analogy to the popular inception module in classical CNNs explains the name of the proposed architecture²¹.
As already mentioned, the matrix products A₁X,…, AᵣX in the above equations do not depend on the learnable model parameters and can thus be pre-computed. In particular, for very large graphs this pre-computation can be scaled efficiently using distributed computing infrastructures such as Apache Spark. This effectively reduces the computational complexity of the overall model to that of an MLP. Moreover, by moving the diffusion to the precomputation step, we can aggregate information from all the neighbors, avoiding sampling and the possible loss of information and bias that comes with it²².
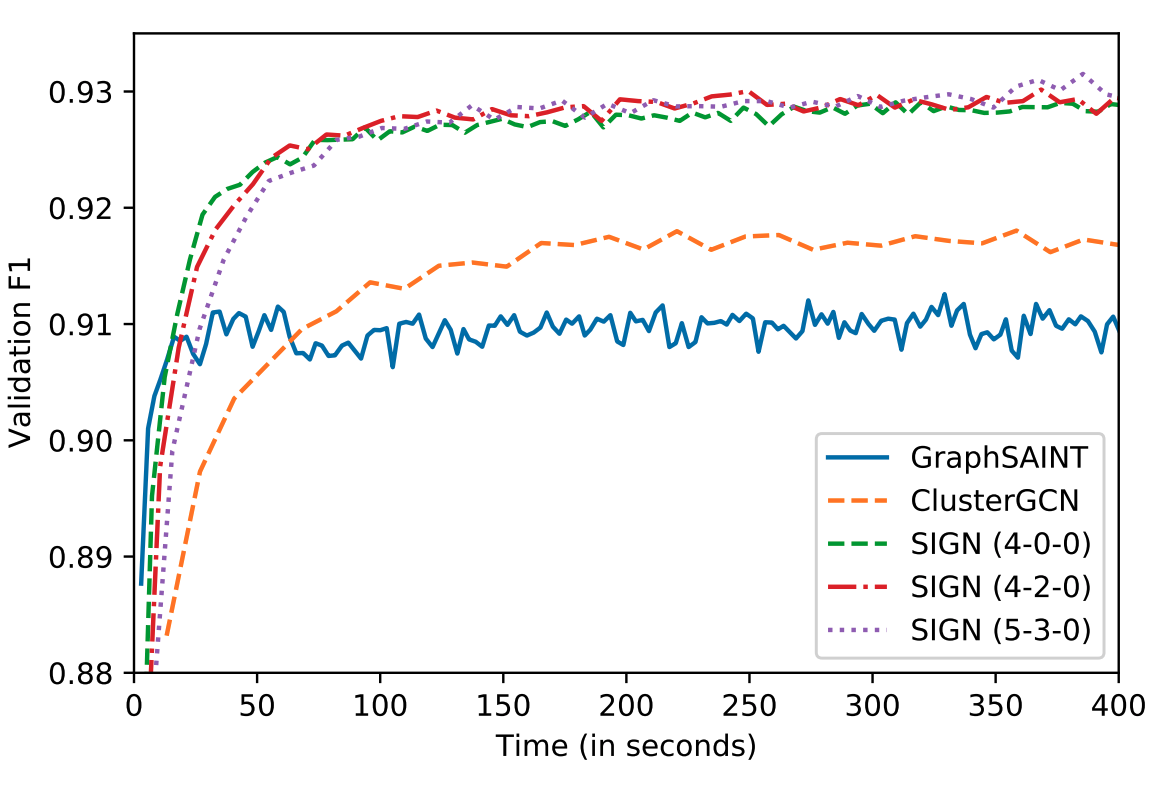
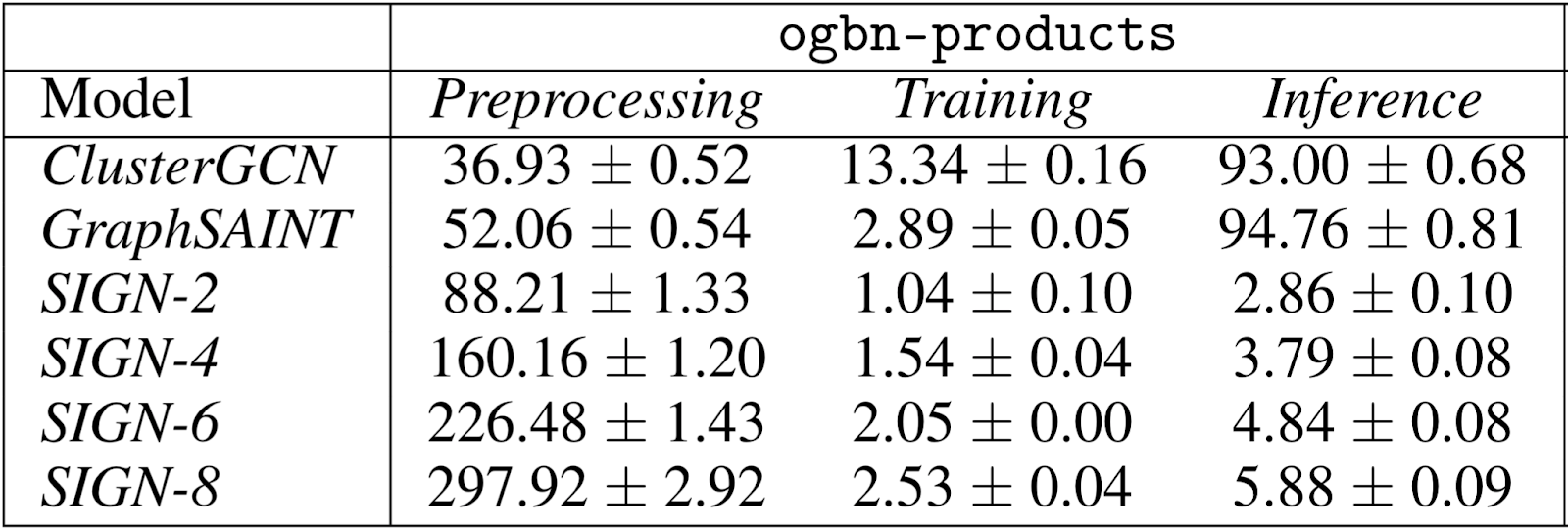
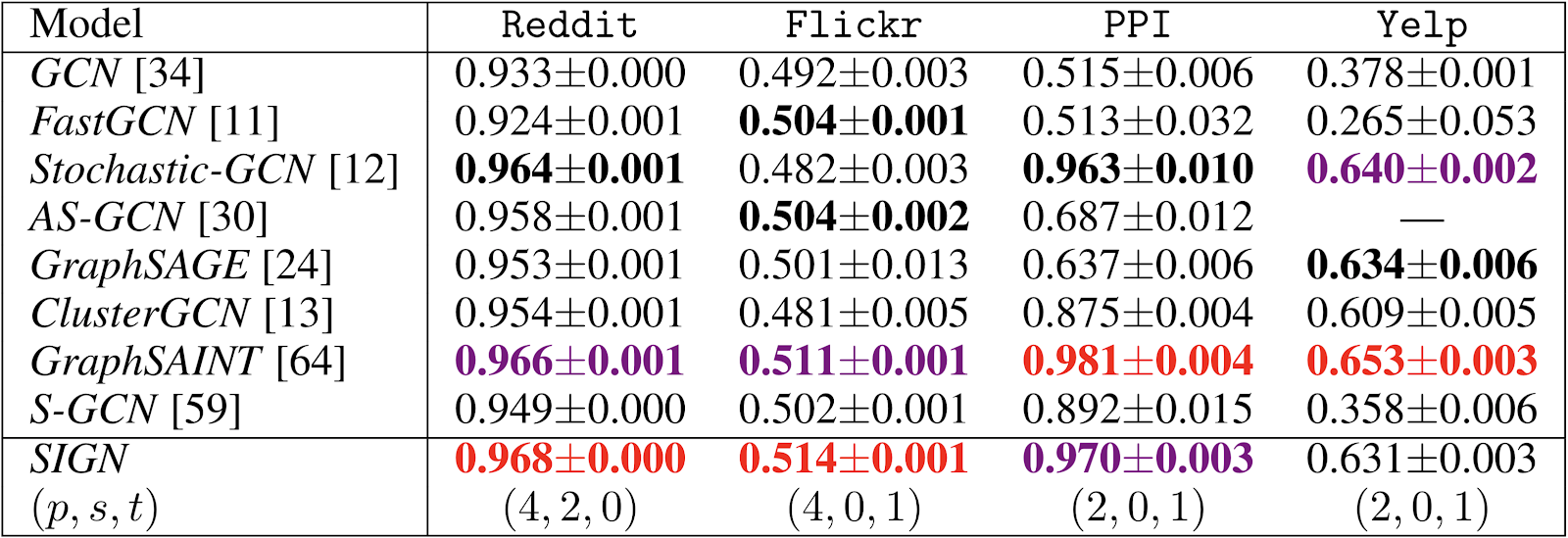
The main advantage of SIGN is its scalability and efficiency, as it can be trained using standard mini-batch gradient descent. Evaluating on popular benchmarks²³⁻²⁴, we found our model to be up to two orders of magnitude faster than ClusterGCN and GraphSAINT at inference time (Table 1). Our model was also significantly faster at training time (Figure 3), while maintaining accuracy performances very close to that of GraphSAINT (Table 2).
In Figure 3, several SIGN variants are reported in the form (p-s-t), with p,s,t indicating, respectively, the maximum employed power of the standard normalized adjacency, Personalized PageRank diffusion, and triangle based operators.
Table 1 shows that while having a slower pre-processing, SIGN is faster at training and nearly two orders of magnitude faster at inference time than other methods. For SIGN-p, the preprocessing step requires p operator applications. In our experiments, the operator applications were computed as sparse matrix multiplications on a single machine. The preprocessing time can potentially be reduced and easily scaled to massive graphs with frameworks for distributed computation, such as Apache Spark.
Moreover, our model supports any kind of diffusion operator. For different types of graphs, different diffusion operators might be necessary and we found some tasks to benefit from having motif-based operators such as triangle counts.
Despite the limitation of having only a single graph convolutional layer and linear diffusion operators, SIGN performs very well in practice, achieving results on par or even better than much more complex models. Recently, SIGN has been successfully applied to the OGBN-Papers100M dataset, the largest publicly available node-wise classification benchmark, with over 110 million nodes and 1.5 billion edges²⁵. Given its speed and simple implementation, we envision SIGN to be a simple baseline graph learning method for large-scale applications. Perhaps more importantly, the success of such a simple model leads to a more fundamental question: “do we really need deep graph neural networks?”
We conjecture that in many learning problems on social networks and “small world” graphs, we should use richer local structures rather than resort to brute-force deep architectures. Interestingly, traditional CNNs architectures evolved according to an opposite trend (deeper networks with smaller filters) because of computational advantages and the ability to compose complex features of simpler ones. We are not sure if the same approach is right for graphs, where compositionality is much more complex. For example, certain structures cannot be computed by message passing, no matter how deep the network is. Surely, more elaborate experiments are still needed to test this conjecture. For now, we invite the reader to take a deeper look at our paper here.
¹ Cora, a citation network containing only about 5K nodes, is still being widely used.The recently introduced Open Graph Benchmark now offers large-scale graphs with millions of nodes. It will probably take some time for the community to switch to it.
² T. Kipf and M. Welling, Semi-supervised classification with graph convolutional networks (2017), In Proc. ICLR introduced the popular GCN architecture, which was derived as a simplification of the ChebNet model proposed by M. Defferrard et al. Convolutional neural networks on graphs with fast localized spectral filtering (2016), In Proc. NIPS.
³ As the diffusion operator, Kipf and Welling employed a symmetrically normalized version of the graph adjacency matrix with self-loops B. That is, A = D-½BD-½ with D the diagonal degree matrix of B. In this case the node itself contributes to its own feature update. Other choices of diffusion operators are possible as well. The diffusion can also be made feature-dependent of the form A(X)X. That is to say, it is still a linear combination of the node features, but the weights depend on the features themselves. This occurs in MoNet⁴, GAT⁵ models. The diffusion can also be completely nonlinear, 𝒜(X), like in message-passing neural networks (MPNN)⁶. For simplicity, we focus the discussion on the GCN model applied to node-wise classification.
⁴ F. Monti et al., Geometric Deep Learning on Graphs and Manifolds Using Mixture Model CNNs (2017), In Proc. CVPR.
⁵ P. Veličković et al., Graph Attention Networks (2018), In Proc. ICLR.
⁶ J. Gilmer et al., Neural message passing for quantum chemistry (2017). In Proc. ICML.
⁷ Here we assume for simplicity that the graph is sparse with the number of edges |ℰ|=E=𝒪(n). In this case, the E term in the memory complexity is “outshined” by the Lnd one. That is, the memory to keep the connectivity into memory becomes negligible with respect to the memory required by all node embeddings.
⁸ W. Hamilton et al., Inductive Representation Learning on Large Graphs (2017), In Proc. NeurIPS.
⁹ The number of neighbors in such graphs tends to grow exponentially with the neighborhood expansion.
¹⁰ Sampling with replacement means that some neighbor nodes can appear more than once, in particular if the number of neighbors is smaller than k.
¹¹ W.-L. Chiang et al., Cluster-GCN: An Efficient Algorithm for Training Deep and Large Graph Convolutional Networks (2019), In Proc. KDD.
¹² H. Zeng et al., GraphSAINT: Graph Sampling Based Inductive Learning Method (2020), In Proc. ICLR.
¹³ Y. Rong et al. DropEdge: Towards deep graph convolutional networks on node classification (2020), In Proc. ICLR. An idea similar to DropOut where a random subset of edges is used during training.
¹⁴ U. Alon and E. Yahav, On the bottleneck of graph neural networks and its practical implications (2020), arXiv:2006.05205. Identified the over-squashing phenomenon in graph neural networks, which is similar to one observed in sequential recurrent models.
¹⁵ Frasca et al., SIGN: Scalable Inception Graph Neural Networks (2020), ICML workshop on Graph Representation Learning and Beyond. The arXiv version is available here.
¹⁶ O. Shchur et al. Pitfalls of graph neural network evaluation (2018). Workshop on Relational Representation Learning. Shows that simple GNN models perform on par with more complex ones.
¹⁷ F. Wu et al., Simplifying graph neural networks (2019). In Proc. ICML.
¹⁸ While we stress that SIGN does not need sampling for computational efficiency, there are other reasons why graph subsampling is useful. J. Klicpera et al. Diffusion improves graph learning (2020), Proc. NeurIPS shows that sampled diffusion matrices improve performance of graph neural networks. We observed the same phenomenon in early SIGN experiments.
¹⁹ G. Bouritsas et al. Improving graph neural network expressivity via subgraph isomorphism counting (2020). arXiv:2006.09252. Shows how provably powerful GNNs can be obtained by structural node encoding.
²⁰ F. Monti, K. Otness, M. M. Bronstein, MotifNet: a motif-based graph convolutional network for directed graphs (2018). arXiv:1802.01572. Uses motif-based diffusion operators.
²¹ C. Szegedi et al., Going deeper with convolution (2015). Proc. CVPR proposed the inception module in the already classical Google LeNet architecture. To be fair, we were not the first to think of graph inception modules. Our collaborator Anees Kazi from TU Munich, who was a visiting student at Imperial College last year, introduced them first.
²² Note that reaching higher-order neighbors is normally achieved by depth-wise stacking graph convolutional layers operating with direct neighbors. In our architecture this is directly achieved in the first layer by powers of graph operators. Details about this dataset can be found here.
²³ More details on the OGBN-Products dataset can be found here; the dataset is made available under the ODC Attribution License.
²⁴ Further details on these benchmarks can be found in GraphSAINT: Graph Sampling Based Inductive Learning Method¹². Instructions for their download, along with the official implementation of the model, can be found at this link.
²⁵. More details on the OGBN-Papers100M dataset can be found here, which is made available under the ODC Attribution License . The public leaderboard is available here.
Did someone say … cookies?
X and its partners use cookies to provide you with a better, safer and
faster service and to support our business. Some cookies are necessary to use
our services, improve our services, and make sure they work properly.
Show more about your choices.